Analysis of Highlights from Health Canada’s GCP Inspection Results in 2016
Publication date: June 2017 / Download as PDF
Author: Irina Mosesova
Introduction
At Vantage BioTrials (VBT) we pride ourselves at continuously learning and keeping up to date with the latest developments in clinical research and compliance issues that can potentially effect trial results or even patient safety. The industry is changing at an incredible rate and in an effort to continue being one of the best service providers for clinical trial management, latest regulatory findings are always on our radar in hopes that we can learn from them and continuously improve our processes and keep patient safety as a forethought rather than an afterthought. In this paper we take a closer look at the top five violations that are particularly of interest. Based on our inquiry to Health Canada, the detailed inspection reports for these non-conformities are not available to the public as they belong to the sponsors of each study, hence we will discuss how these violations fit into the historical data that is available, such as Annual Inspection Summary Reports. We will also look at how these trends have contributed and resulted in the evolution of regulatory guidelines and regulations.
According to the Government of Canada (2), eight inspections of investigative sites participating in clinical trials in 2016 resulted in non-compliant ratings. Figure 1 – Frequency vs. Summary of Observations by Health Canada GCP Inspectors in 2016 illustrates which non-conformities were the most frequent, with the top six highlighted in red below.
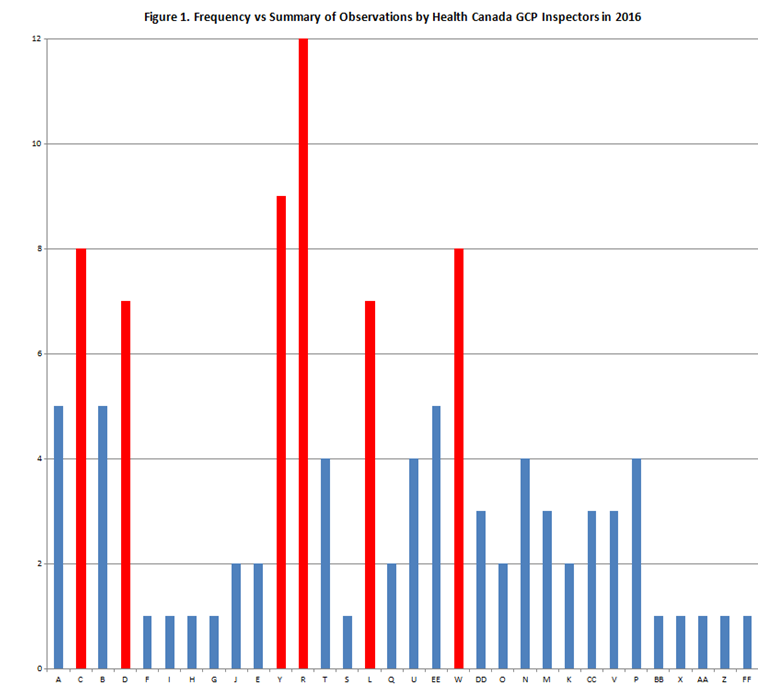
Table 1. Summary of Observations noted by GCP Inspectors in 2016 with their corresponding Codes.
| Code | Summary of Observations |
| A | Medical care and/or medical decisions for the clinical trial were not under the supervision of the qualified investigator at the clinical trial site. |
| B | Not all individuals conducting the clinical trial had the education, training and experience to perform their respective tasks. |
| C | The clinical trial records had errors and/or missing information that did not allow for complete and accurate reporting, interpretation, and verification. |
| D | The clinical trial was not conducted according to the protocol. |
| E | The drug was not handled and stored in keeping with Good Manufacturing Practices. |
| F | The drug was sold or imported for use in a clinical trial before the clinical trial site information was submitted to Health Canada. |
| G | The label of the drug did not contain the required information. |
| H | The sponsor did not get the approval of a Research Ethics Board before the clinical trial began or before an amendment was made at the clinical trial site. |
| I | The sponsor did not get written informed consent for every person before they participated in the clinical trial or the amended clinical trial. |
| J | The sponsor did not have provisions in place to keep all clinical trial records for a period of 25 years. |
| K | The sponsor did not implement systems and procedures to ensure accurate and complete transcription of data. |
| L | The sponsor did not implement systems and procedures to ensure adequate monitoring of the clinical trial. |
| M | The sponsor did not implement systems and procedures to ensure adverse reactions or events were documented and reported. |
| N | The sponsor did not implement systems and procedures to ensure equipment was maintained and calibrated. |
| O | The sponsor did not implement systems and procedures to ensure that deviations from the protocol were documented and reported. |
| P | The sponsor did not implement systems and procedures to ensure that participants were properly consented. |
| Q | The sponsor did not implement systems and procedures to ensure that staff was adequately trained on Good Clinical Practices and the appropriate Food and Drug Regulations. |
| R | The sponsor did not implement systems and procedures to ensure the quality of the clinical trial. |
| S | The sponsor did not implement systems and procedures to ensure the required information was submitted to the Research Ethics Board. |
| T | The sponsor did not implement systems and procedures to ensure the required study information was communicated to and/or reviewed by appropriate study personnel in a timely manner. |
| U | The sponsor did not implement systems and procedures to ensure the study records were maintained and archived. |
| V | The sponsor did not implement systems and procedures to ensure the tasks were appropriately delegated to study staff. |
| W | The sponsor did not implement systems and procedures to train study staff. |
| X | The sponsor did not keep complete and accurate records for the use of the drug in a clinical trial, as required by law. |
| Y | The sponsor did not keep complete and accurate records to show the clinical trial was conducted in keeping with Good Clinical Practices and the Regulations. |
| Z | The sponsor did not keep records of all adverse events. |
| AA | The sponsor did not meet the requirements for information and records management. |
| BB | The sponsor did not notify Health Canada within 15 days of making a change to the chemistry and manufacturing information of the drug that does not affect the quality or safety of the drug. |
| CC | The sponsor did not notify Health Canada within 15 days of making a change to the protocol that does not alter the risk to the health of a clinical trial participant. |
| DD | The sponsor did not record, handle and store all information for a clinical trial to ensure the data transcribed from the original documents to case reports was accurate and complete. |
| EE | The sponsor did not record, handle and store all information for the delegation of tasks in the clinical trial to allow for complete and accurate reporting, interpretation, and verification. |
| FF | The sponsor did not implement systems and procedures to ensure the quality of the clinical trial. |
C.05.012 – Records
The clinical trial records had errors and/or missing information that did not allow for complete and accurate reporting, interpretation, and verification.
The sponsor did not keep complete and accurate records to show the clinical trial was conducted in keeping with Good Clinical Practices and the Regulations.
This is quite a large category that arguably could be considered as the backbone of the regulations. If you cannot trust your records, then everything becomes questionable. This section of the Food and Drug Regulations (C.05.012) was sited 30% of the total number of observations during GCP inspections in Canada between 2014-2015 (7). The violations that fall under these categories were also seen in a 2012 report published by Health Canada that summarized the inspections between April 2004 and March 2011 (3). In that report, electronic records seem to be a recurrent non-conformity. Given the fast-paced integration of technology into clinical research, it is surprising how much confusion is still out there when it comes to the use of computerized systems and collection of e-data. Regulatory agencies have recognized the need to address and provide guidance for the use of computerized systems in clinical research, which resulted in several publications of guidelines and regulations to address these issues. For example, in 2010 the EMA published a reflection paper that heavily discussed e-TMFs (4). Another good example is the most recent updates to the ICH E6 guidelines (Revision 2) that has included a new section on validation of computerized systems (6). Furthermore, Health Canada inspectors stated in the Annual Inspection Summary Report that “As of 2014-2015, inspections have become more in-depth and now include the review of more systems (such as the establishment’s self-inspection program and validation of computerized systems).” (7).
The examples in Health Canada’s 2012 report (3) refer to the lack of validation of computerized systems and electronic forms developed in-house and used in a clinical trial. During one of the inspections they also noted the lack of procedure in place for archiving the electronic records for the required period of time. Many still fail to recognize that electronically captured data is raw data and should be treated with the same scrutiny as paper-based data, i.e. be compliant with FDA CFR 21 Part 11 or equivalent regulations. Interestingly, one of the examples of the most common observations in the annual 2014-2015 Health Canada report also lists lack of appropriate systems and procedures in place for computerized system validation but these were flagged under section C.05.010(c) instead (7).
The industry is recognizing the importance of regulated software and more software development companies are beginning to claim their products as compliant with appropriate regulations. However, caution is advisable: don’t simply take their word for it. Do your homework and (a) conduct a vigorous vendor assessment and, (b) perform a full or partial in-house software validation to ensure that the software is fit-for-use in your operational environment
C.05.010 – Sponsor’s Obligations
Good Clinical Practices – The sponsor did not implement systems and procedures to ensure adequate monitoring of the clinical trial.
As a Monitor (i.e. Clinical Research Associate) it is essential to keep in mind that we play a critical role in ensuring compliance in any particular clinical study. The following interesting example from FDA serves as a great reminder of this fact.
Last year, Dr. Cheta Nand’s site was inspected by the FDA in Richland, Washington, USA (1). This inspection resulted in a Warning Letter with six violations cited. In one of the findings, FDA inspectors noted that the repeat of one of the laboratory parameters was not performed when elevated levels were noted in the original results, as required by the protocol. Interestingly, one of the justifications that Dr. Nand offered to the FDA, which was deemed unacceptable, is that his study coordinator received “a verbal confirmation from the study monitor” that this was an acceptable course of action. In fact, this justification was used repeatedly in response to several findings. Moral of the story: what we do and say as Monitors matters and can, under some circumstances, be used to justify the violations. It is also worth noting that verbal communications are weak evidence to justify anything and the Golden Rule of clinical research “if it’s not documented, it didn’t happen” should not be taken for granted.
Given the repeated violation of the sponsor’s oversight in regard to the monitoring of the clinical trial, it’s no wonder that a significant portion of the original ICH E6 guideline was recently revised to include the discussion on Risk Based Monitoring, Monitoring Plans, and Monitoring Reports (6). This was great news to all, especially our team at Vantage BioTrials, given that we have been implementing this process since our inception ten years ago in 2007.
Good Clinical Practices – The sponsor did not implement systems and procedures to ensure the quality of the clinical trial.
The ultimate responsibility of ensuring the quality of the clinical trial falls on the sponsor but every single person involved plays an important role. It is a dynamic and continuous process. According to the FDA’s presentation – Building Quality Into Clinical Trials – An FDA Perspective (5), ”quality” means that the study needs to have the following elements: scientifically valid and ethically sound experimental design, adequate protection of subjects’ rights, safety, and welfare, qualified personnel, adequate monitoring, and current, complete, and accurate data. FDA suggests the following items to be considered for a quality study:
- Adequate effort is spent in finding a qualified investigator. Solely relying on Investigator’s CV and the reputation is not sufficient. It’s important to also pay attention to the clinical site and staff, evaluating the understanding of the appropriate regulatory documents, considering skills and experiences with similar products, etc.
- Adequate effort is put into writing the protocol, which might mean spending more time and effort at the start-up of the study to avoid confusion and protocol deviations in the future. Getting a proper input from the relevant parties prior to finalizing the protocol is the recommended strategy for any clinical trial.
- Adequate and thorough training, with emphasis on the informed consent process.
- Ensuring adequate monitoring and investigator’s compliance throughout the study.
- Ensuring all third parties involved on the study also comply with the appropriate regulations.
For further examples and practical tips on Quality by Design Principles, feel free to refer to our Whitepaper entitled “Quality by Design and Risk-Based Monitoring” downloadable through our website at vantagebiotrials.com.
Good Clinical Practices – The sponsor did not implement systems and procedures to train study staff.
In the author’s ten years as an auditor, training has always been a hot subject to highlight by sponsors and regulatory agencies. Having competent and qualified staff is the foundation of any business providing a niche service, yet somehow adequate and timely training is a constant oversight and almost a guaranteed violation during site audits.
Health Canada’s annual report for 2014-2015 noted that training was amongst the top three observations (7). This was also noted in Health Canada’s report published in 2012, which included examples of deficiencies cited again in this section. Below are two examples that are particularly interesting:
“Example 1: Lack of documented evidence that the personnel, including study coordinators, involved in the conduct of the study were trained on the protocol and GCPs.”
The most common challenge noted in the author’s monitoring and auditing experience is the lack of clearly defined training file ownership. At times, even the physical location of the training file might result in confusion. For example, the training file located in the management’s office can leave an employee under the impression that it’s his or her boss’ responsibility to keep track of all training. A concise training SOP clearly defining responsibilities and ownership of the training files is a must for every operational site.
“Example 2: There was no explicit documentation to indicate that all sub-investigators and nurses to whom significant trial-related duties have been delegated had been informed of all protocol-specific requirements.”
Similar observations were noted in Health Canada’s annual report for 2014-2015 (7).
The author once had a sponsor who argued that the person who wrote the protocol had to also sign off on a “Protocol Reading Log”. This might be an example of overkill and overzealousness, but in all other cases a written attestation to understanding the protocol-specific requirements shows that study management has done their due-diligence, not to mention that an opportunity was provided (and documented) to all involved personnel to ask questions or raise concerns.
It is evident that there is a lot to consider in the process of developing and managing a compliant quality clinical trial. Health Canada noted that in response to the rapid growth of new products coming to market, “Health Canada is developing a suite of new tools to help identify risks and shift from regular inspection cycles to a more flexible risk-based approach.” (7). As a CRO, Vantage BioTrials has built its operational philosophy on remaining lean and flexible, even when implementing risk-based and “quality by design” methodologies since Day One of operations. Not only are we among the pioneers to incorporate these quality methods into our clinical management and monitoring activities, we also deliver highly experienced and qualified professionals that can support your team in every aspect of Phase I-IV drug development. But don’t take our word for it, let our awards and accomplishments speak for themselves!
About the Author
Irina Mosesova holds an advanced B.Sc. degree in Microbiology & Immunology from Dalhousie University. After spending several years at the bench of the MUHC Royal Victoria Hospital discovering novel gene mutations in the patients with deficient calcium-sensing receptor, Irina continued her career in the pre-clinical regulatory affairs field, where she spent over ten years. Last year, Irina’s focus has shifted in hopes to continue her learning journey and in effort to contribute her skills and knowledge to clinical research. She currently holds a position of a Clinical Trial Assistant at Vantage BioTrials located in Montreal, Quebec, Canada.
About us
Founded in 2007, Vantage BioTrials is a privately-held Contract Research Organization (CRO) that provides Phase I-IV clinical trial management services and implements Risk-Based Management through the use of Quality by Design (QbD) methodologies, all delivered to international pharmaceutical, biotechnology, generic pharmaceutical and medical device companies.
To learn more about Vantage BioTrials, please visit us at vantagebiotrials.com and follow us on LinkedIn and Twitter.
References
- FDA Warning Letter to Dr. Cheta Nand, M.D. 3/10/16. Ref. 16-HFD-45-03-01. Website: http://www.fda.gov/iceci/enforcementactions/warningletters/2016/ucm493102.htm
- Clinical trial inspections, non-compliant clinical trial inspections search results. Website: http://healthycanadians.gc.ca/apps/gcp-bpc/searchResult-en.html?lang=en&sponName=&contNum=®ion=&drugName=&phase=&startDate=&rating=NC&cat=2
- Summary Report of Inspections of Clinical Trials Conducted from April 2004 to March 2011. Health and Drug Products. Website: http://www.hc-sc.gc.ca/dhp-mps/compli-conform/clini-pract-prat/report-rapport/2004-2011-eng.php#a53
- Reflection paper on GCP compliance in relation to trial master files (paper and/or electronic) for management, audit and inspection of clinical trials. Website: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/02/WC500138893.pdf
- Building Quality Into Clinical Trials – an FDA Perspective by Jean Toth-Allen, PhD, U.S. Food and Drug Administration. Website: https://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/SmallBusinessAssistance/UCM303954.pdf
- Integrated Addendum to ICH E6(R1): Guideline for Good Clinical Practice E6(R2).ICH. Website: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R2__Step_4.pdf
- Inspectorate Program – Annual Inspection Summary Report 2014-2015. Health and Drug Products. Website: http://www.hc-sc.gc.ca/dhp-mps/pubs/compli-conform/2014-2015-ar-ra/index-eng.php